
|
If this is your first contact with DireX, read some General Remarks first. The input files needed to run the tutorials can be downloaded here: direx-0.4-tutorial.tgz There is one folder for each tutorial. Each folder contains an executable script file (run.sh) which runs through all the steps. Open this script in a text editor to modify it and to learn how to run DireX. For all tutorials that involve simulated density maps, the maps are generated by the run.sh script as well. This allows you to easily run the test cases at different resolutions, if you like. |
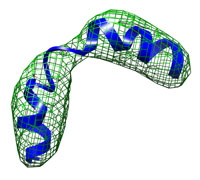
| A simple toy example: A small kinked helix at low resolution (10 Å) runtime = 12 sec number of atoms = 206 This tutorial shows the basic usage and demonstrates how the DireX/DEN approach allows to describe breakage of secondary structure elements. Start here... |
|
|
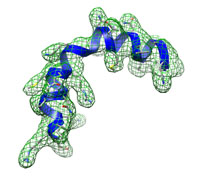
The small kinked helix at high resolution (3 Å) typical runtime = 3 min number of atoms = 206 DireX can be used to assist model building by real-space refinement also at high resolution. Start here... |
|
|
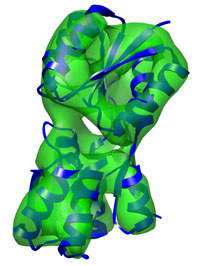
Refinement of the Ribose-binding Protein, a rather simple
two-domain protein typical runtime = 2 min
number of atoms = 2465 Basic DireX usage for a refinement of a protein at 10Å showing a pure hinge motion. Start here... |
|
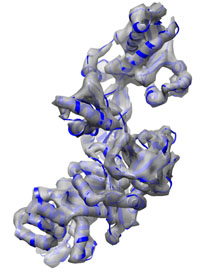
| Somewhat more complicated: Elongation Factor 2 typical runtime = 1 h number of atoms = 6376 Two crystal structures with an RMSD of about 15 Å are used as starting and target structure. Refinement using a 8 Å density map leads to a final RMSD of only 0.6 Å. This tutorial demonstrates how to use secondary structure information and Tirion enhanced sampling to cover larger conformational changes. Start here... |
|
|
Resolving atom clashes: How to deal with overlapping composite models typical runtime = 3 min number of atoms = 3062 Building a composite model consisting of several proteins/subunits often results in some atomic overlap. This often leads to problems and should be resolved before the flexible fitting is performed. Here is described how to do this. Start here... |
|
|
Occupancy refinement: typical runtime = 24 sec number of atoms = 402 Flexible parts of a protein usually show reduced or even missing density in the experimental map. This effect can lead to significant distortion in the refined structure. DireX uses the occupancy refinement feature to account for reduced density which typically improves the fit. Start here... |
|
|
Application to real data: GroEL at 6 Å This example shows the usage of B-factor/Occupancy refinement and usage of NCS restraints (for multiple idential subunits). NOT YET AVAILABLE ! Start here... |
|
|
Computing Pathways of Conformational Transitions:
Myosin
DireX can be used to find possible pathways of conformational transitions. This example does not involve any density maps. NOT YET AVAILABLE ! Start here... |
|