
|
DireX - Tutorial Somewhat more complicated: Elongation Factor 2 |
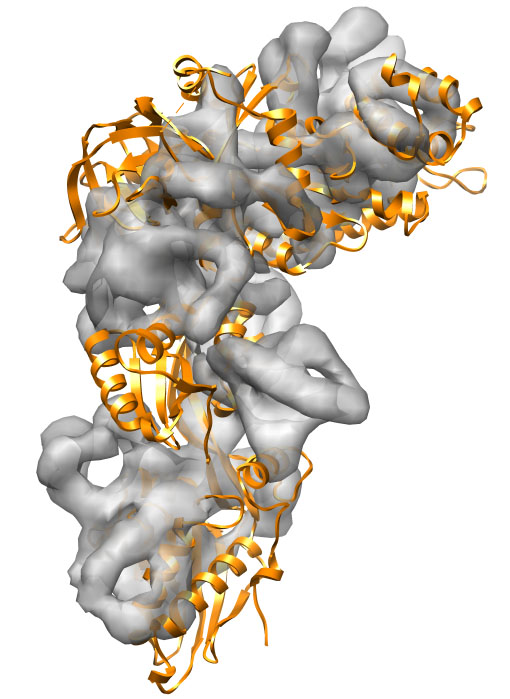
Starting from the 1n0u PDB structure of the Elongation Factor 2 the goal is to get as close as possible to the 1n0v PDB structure, from which a density map is generated at 8 Å. The RMSD between both structures is about 13 Å. Since the calculation takes about one hour, we provide the results (in the ./results/ folder).
As we have seen earlier, the refinement is performed in two steps (see run.sh): A first round with larger sampling steps
direx -pdb start-1n0u.pdb -map ef2-density.mrc -f refine.par -ox traj-refine.xtc -op out-refine -curmap curmap-refine.mrc -cur current-refine.pdb -mapcc mapcc-refine.dat -secstr 1n0u.ss
followed by a minimization-like calculation with small step sizes (see below).Tirion-enhanced Sampling
For the first round we use a feature in DireX called 'Tirion-enhanced sampling' which allows larger conformational motions per iteration. This feature is activated by setting tirion_use = yes in the parameter file.
Tirion-enhanced sampling uses a smarter and more collective perturbation of the coordinates than pert_fac. This basically performs a nested CONCOORD run only on Cα positions to determine a perturbation vector for each residue. All atoms within the same residues are then moved along this perturbation vector which leads to less local distortions.
If you have a very smooth density map and you do not want to sample a large conformational space, you probably will not need this. But in many cases experimental density maps have a lot of noise and if you want to sample many conformations (e.g. at low resolution), then the Tirion-enhanced sampling can improve the refinement and speed up the sampling a lot.
Two parameters are important here:
tirion_pert_fac: Initial gaussian perturbation of the Cα positions.
tirion_fac: Scales the stepsize along the perturbation vector determined by the nested CONCOORD run.
If you want to increase the stepsize between two structure generation steps, you only need to increase tirion_fac.
Use of Secondary Structure Information
Loop regions are typically much more flexible than α-helices or β-sheets. This knowledge can be used to further tune the strength of the DEN restraints. The parameter den_loop scales those DEN restraints that are within loop elements or between any other element and a loop element. The secondary structure assignment is read with the option -secstr. The file contains five columns and needs to be in this format:
Number ResID ChainID Single-letter_Aminoacid Secondary_Structure_Code
The secondary structure code (H,E,G,T,etc) is the same as is used by the program DSSP. As an example (excerpt from 1n0u.ss):
[...]
100 119 A L H
101 120 A R H
102 121 A V T
103 122 A T
104 123 A D S
105 124 A G E
106 125 A A E
[...]
When reading a structure with the -pdb option only, then those coordinates are used for selecting the DEN restraints and are also taken as the reference coordinates.
When using the -pdb file1.pdb option and the -p file2.pdb option, then only topology and geometry (bond definitions and bond lengths and angles) are read from file1.pdb, the coordinates are however taken from file2.pdb. In this case the file2.pdb coordinates are used to select the DEN restraints and also as the reference coordinates.
The -refden file3.pdb option can be used to read reference coordinates in case you want them to be different from file2.pdb.
A list of all DEN restraints can be written to a file by using the option -oden den.dat. The file den.dat then contains for each DEN restraint the id of the two atoms that are restraint, the reference distance and the current equilibrium distance of the restraint.
The -iden den.dat option is used to read in a list of DEN restraints.
In our specific case, we do:
# Prepare DEN restraints for final minimization.
direx -pdb start-1n0u.pdb -p current-refine.pdb -f mkden.par -cur current-mkden.pdb -oden oden-refined.dat -refden start-1n0u.pdb
The DEN restraints file is then read with the -iden option and the final step is performed:
# Final refinement with only small perturbations.
# This is almost like a minimization.
direx -pdb start-1n0u.pdb -p current-refine.pdb -map ef2-density.mrc -f min.par -ox traj-min.xtc -op out -curmap curmap-min.mrc -cur current-min.pdb -mapcc mapcc-min.dat -secstr 1n0u.ss -iden oden-refined.dat
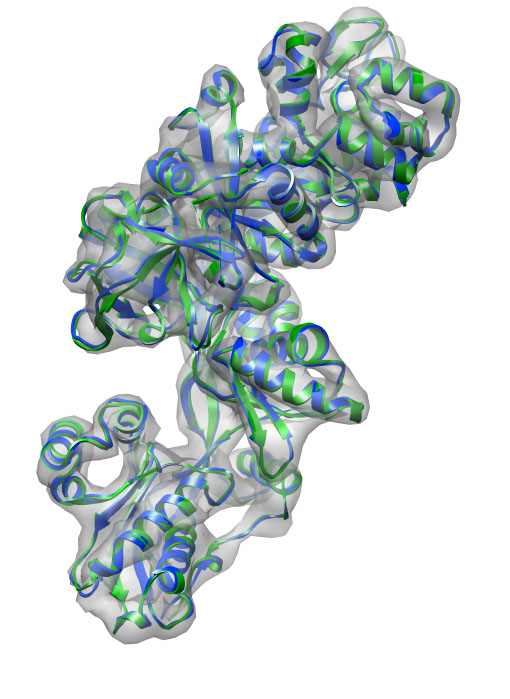 The structure current-min.pdb contains the final fitted structure.
The structure current-min.pdb contains the final fitted structure.
Analysis
The final RMSD is about 0.6 Å. As always, you can watch the trajectory with VMD:
vmd -f start-1n0u.pdb ./results/traj-refine.xtc ./results/traj-min.xtc -m ./results/ef2-density.mrc target-1n0v.pdb