
Conformational Sampling Algorithm
DireX uses the CONCOORD algorithm (see de Groot, et al., Proteins 29: 240-251 (1997)) to perform the conformational sampling moves. The CONCOORD algorithm is applied iteratively to obtain a trajectory of structures that progressively sample conformational space.
|
In each step of this iteration CONCOORD defines a large number of distance
restraints which are represented as allowed distance intervals.
There are two types of retraints: 1) topological restraints, which ensure that the
model retains correct stereochemistry, and 2) van der Waals restraints, which prevent
atom overlaps and set an upper limit for the allowed conformational change.
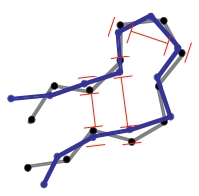
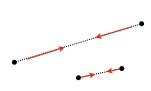
In a next step the coordinates of the structure (gray) are randomly perturbed by using Gaussian distribution with a width that is set by pert_fac in the parameter file. Then, the coordinates are iteratively corrected to fulfill the CONCOORD restraints (red) and to eventually produce a new structure (blue). This correction cycle is the core of the CONCOORD algorithm, which traverses the list of CONCOORD restraints in random order, and corrects those disances that lie outside their allowed interval. For each violated restraint, the two corresponding atoms are moved along their interatomic vector toward a target distance, which is randomly picked from within the allowed interval of the particular restraint. |
|